Risk Diagram: Part 1- Latent level of Risk
Analysing the latent level of risk in medical device, combination product development. (originally published as part of my LinkedIn newsletter)
Noel Butterworth
2/18/20268 min read
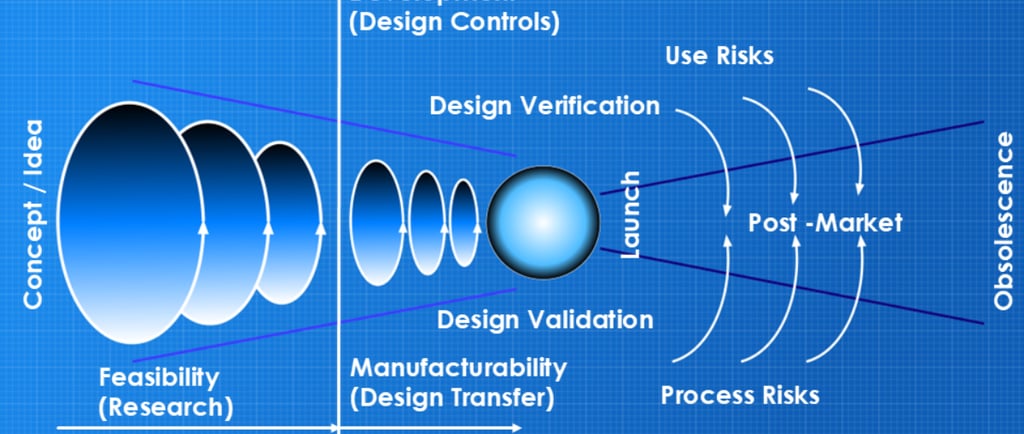
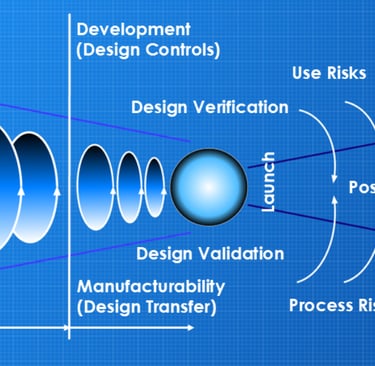
The original 2016 version of the risk diagram
Back in 2016, I put together a diagram that would explain the latent level of risk in medical device development*, mapping it relative to the requirements of the quality process called ‘Design Controls’. I’ve used this diagram in numerous presentations at forums and conferences, whilst encouraging my teams to apply this methodology with successful results. In 2024, I updated the diagram to align with the latest regulatory and International Standards requirements by adding more detail; however, the principle remained the same - to show the management of the latent level of risk.
Firstly, the use of the word ‘risk’ here needs some explanation before I incur the wrath of my numerous Risk Management expert associates. In April 2016, I presented at the Medtec Europe trade fair in Stuttgart on “Practical Guidance to Design, Development and Manufacture of Devices” and began with a comparison of risk to ‘The Force’ in Star Wars; it surrounds us, it defines the galaxy, it permeates through board games.


Slide from my 2016 Medtec Europe presentation “Practical Guidance to Design, Development and Manufacture of Devices”
Here, ‘risk’ is the more literal dictionary definition of the word, and referencing my battered old Oxford English Dictionary from 1990 being defined as “n. 1. The possibility of meeting danger or suffering harm or loss: exposure to this”.


My battered old and still reliable 1990 copy of the OED!
Any medical device being launched for patient use has a latent level of ‘risk’ (dictionary definition). In fact, the device that has zero risk to patients is the device that is not released to market. One contributing factor to determine that the product may not, should not, must not be released to market is the Benefit-Risk assessment, with the Risk (capital ‘R’) calculated as per International Standard ISO 14971:2019 ‘Medical Devices Application of risk management to medical devices’ (and we’ll deep-dive on this terminology in future articles).
The level of risk I was considering in 2016 was a more general risk than the Risk of ISO 1491. In project management, there are additional project/program risks determined and discussed, not to be confused with the ISO 14971:2019 definition. E.g.; financial risks, company profile risks, etc., for failures of a product in the field. These being balanced with the inherent risks coming from the design, manufacture, and/or use of the product (individually or systemic combinations therein) from a project management perspective.
For example, there’s a misconception that the requirement of the Design Control process’s Design Transfer phase is to be completed after Design [and Development] Validation. This is not a stated requirement of the prior FDA’s QMS nor the current ISO 13485:2016 ‘Medical devices - Quality management systems’ section 7. The requirement for Design Validation to be on production processes or equivalent; the discussion begins with the definition of ‘equivalent’. For a product that has been produced in small scale to allow verification and validation testing, there is often concern from project or program management for the potential financial cost to scale up the production process prior to the design having been verified and validated, thus the financial and timescale risks therein.
This is a circular concern due to the potential risk that the production scale-up activities could cause a design specification change, invalidating verification or validation testing. For example, a plastic injection-moulded component that does not achieve its required process capability Ppk or Cpk requirements may need the specification to be widened— specifically the tolerance range of the specification. That would have a knock-on effect for the specification to be assessed for the new tolerance range, which would be a new verification activity.
Hence the emphasis is on the definition of an “equivalent production process” and the assessment of changes through the scale-up activities. Design Transfer planning has to capture all these scenarios.
The requirements of International Standards ISO 13485:2016 and ISO 14971:2019 by definition apply throughout the lifecycle of the device. Or ‘life cycle’, or ‘life-cycle’, per the three different ways it’s written in those two standards plus the additional guidance document ‘Medical devices - A practical guide’ which, from a Quality Manager perspective, is one of those observations that gets under the skin.
However, significantly, lifecycle (I’m picking that one) applies from Initial Conception and hence the mapping diagram I defined likewise begins from Initial Conception (or Concept / Ideation), through the pre-market development, through product launch and through to post-market and obsolescence.
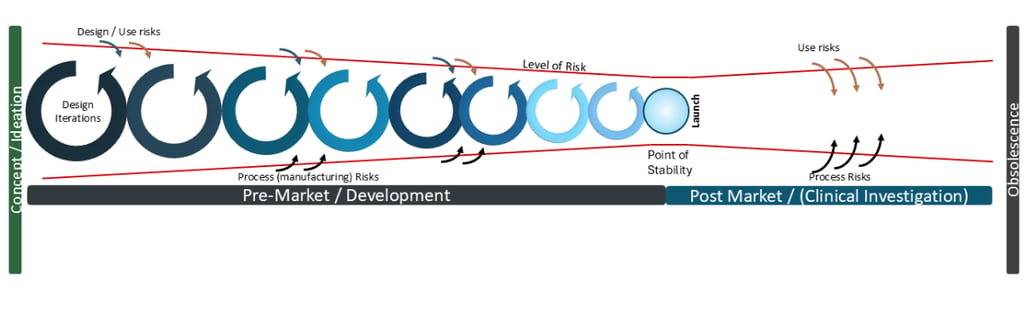
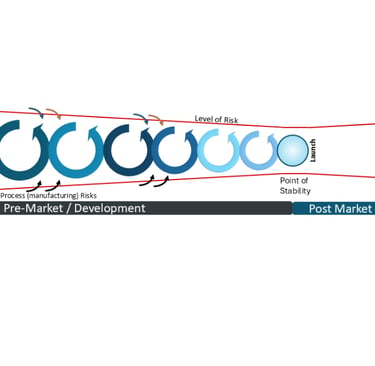
The Level of Risk throughout product lifecycle- pre and post market launch
The inherent level of risk reduces throughout the development cycle until it reaches a Point of Stability whereby the product is deemed sufficiently safe and effective to reach patients. That point is a hugely significant point of determination and we will deep-dive on this significance and utilisation for post-market risks and potential changes in future articles.
There are two main areas of post-market focus:
1) Use risks
Despite the effort during development, use risks are subjective and humans are humans— there will always be someone using your product in a way that could not have possibly been foreseen.
At this point, I will note that the diagram was biased to my personal experience of medical device development, ie hardware. However, and I will reflect on this in coming articles, I believe the principles also apply to software development and will give comparative statements where applicable.
2) Process risks
The other element of latent post-market risk, for hardware specifically, is changes to the manufacturing process. Changes can be required for good reasons, such as a successful product requiring a larger capacity of manufacture than planned; more equipment, larger (or new) facilities, etc. But having 100 CNC machines of the same make and model does not necessarily guarantee that they will all work exactly the same.
The root cause for this is because “anything engineered (designed and manufactured) is unique - and has a propensity to fail”. It’s the consequence of entropy as defined by the Second Law of Thermodynamics and the reason why time (currently) moves in solely one direction, to be profound.
Anything engineered is unique - and has a propensity to fail
Hence, management of risk is a never-ending beast, regardless of the effort in development. Notably, though, I do not flag design as a cause of post-market risk, per the diagram. This is where we add the layer of Design Controls (as defined by ISO 13485:2016 section 7 and was defined by the QMS 21 CFR 820 part 30) to the diagram, with activities leading to the Point of Stability: Design [and Development] Verification plus Design [and Development] Validation. Thus, any and all post-market changes must circle back through this process and be verified and validated, or determine the changes make zero impact on the existing verification, validation to bring the product back to the Point of Stability.
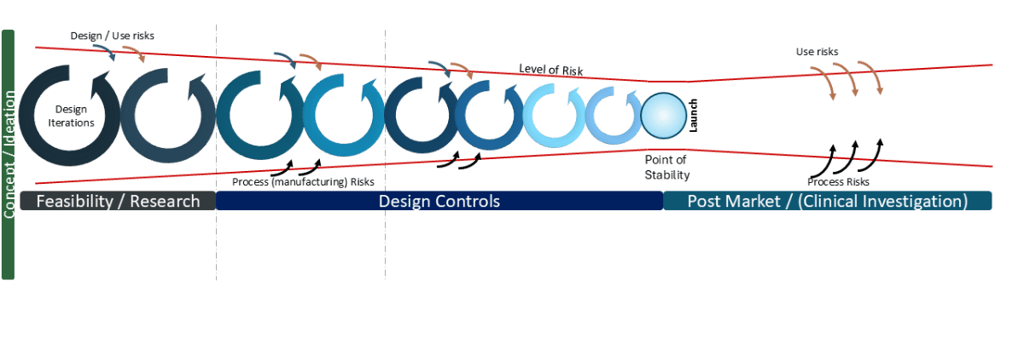
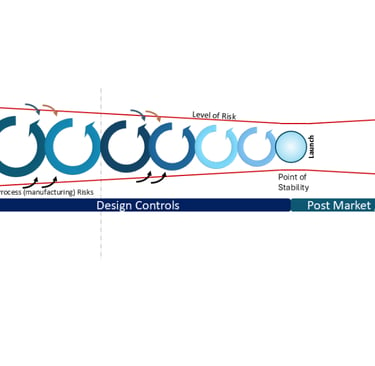
Risk Diagram relative to Design Controls (simplified)
Any design changes as a result of user feedback must circle back through this process. Any design changes as a result of production process changes must circle back through this requirement. Hence, all manufacturing process changes must be risk-assessed that they have no impact on the function of the product (ie changing design specifications), otherwise, that requested change must also circle back through this process.
For software, (per the requirements of IEC 62304:2006 ‘Medical device software - Software life cycle processes’) any software configuration changes must circle back through this process and be determined to either not impact verification and validation, or be reverified, revalidated, etc. For software running on hardware, there must be continual assessment of hardware changes having a net impact and inherent risk to the functioning of the software— equivalent to process changes described above. AI being used with medical device software is a particularly interesting case of how to manage post the Point of Stability, due to risks of hallucinations, data drift, etc.
When I first mapped this diagram in 2016, the focus was on the latent level of risk relative to the Design Control process. The reduction in level of risk aligned with a principle of Design ‘funnelling’ which I first learned of from consultancy PDD Group LTD and their presentation “Insight, Innovation & Design” back in 2007. Over the time of development, the size of design change iteration should reduce, ie in early phase prototyping the amount of changes should be significantly more than minor refinements occurring at the end of the development. Put another way, it’s a failure in development if in the period prior to a planned launch, there’s a sudden requirement for a major design change (potentially caused by manufacturing feedback or potential failures in Design Verification or Design Validation testing). Ie a large design iteration loop raises the latent level of risk again.

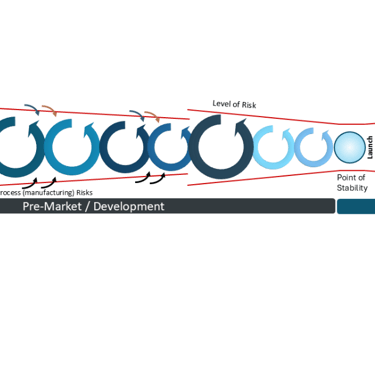
A late in development major design change has a high level of latent risk
Notably, in recent years I’ve seen numerous diagrams for Agile Project Management with similar reducing loops of iterations, aligned to reductions in sizes of “sprints”. Next time, we’ll look more closely into design iterations.
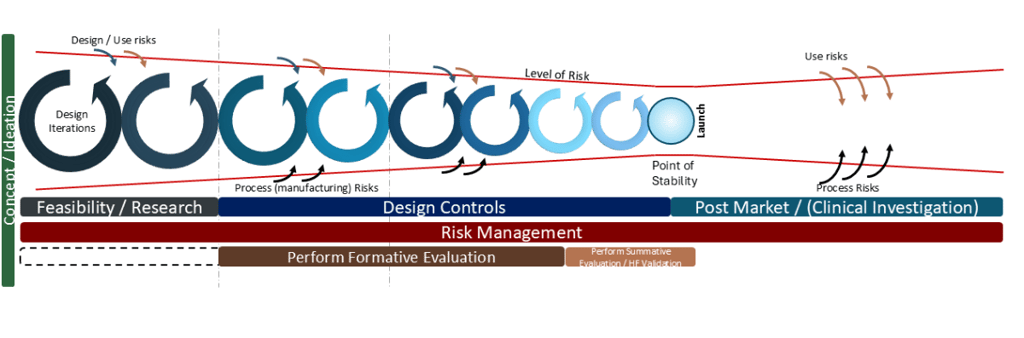
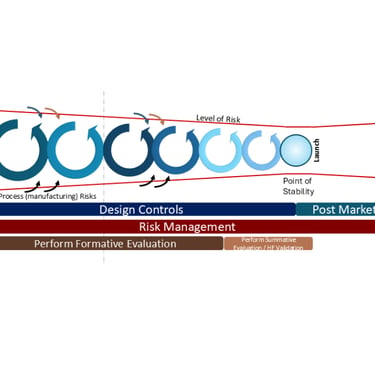
Adding the next level of detail- Risk Management and Usability / Human Factors
When the FDA announced their transition to the ISO 13485:2106, the QMSR which became effective this past 2 February, there was a notable emphasis on the importance of Risk Management. I updated my diagram to include an extra level of detail related to the requirements as defined by ISO 14971:2019 and chose to include the alignments with requirements of Usability and Human Factors, primarily aligned to IEC 62366-1:2015 ‘Medical devices - Part 1: Application of usability engineering to medical devices’. The level of detail worked well for the “big screen” as per my presentation at the ‘International Conference on Medical Device Safety Risk Management’ in Amsterdam last April. However, for the purposes of this newsletter I’m rebuilding the diagram and, much like a subscription service for a magazine that you can build a model of the Titanic, the future articles will build the diagram “step-by-step” to the detail I’ve previously presented, with greater focus and examples.
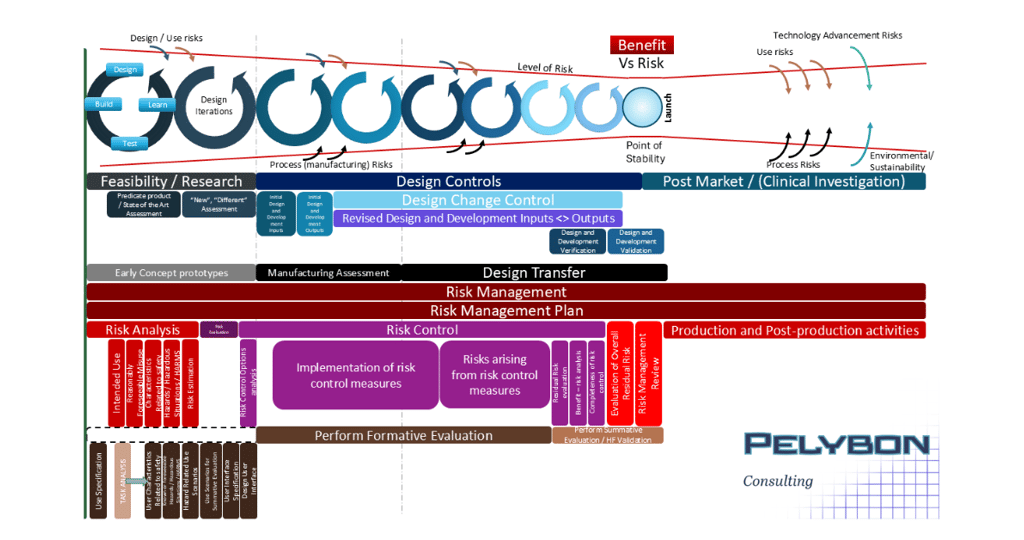
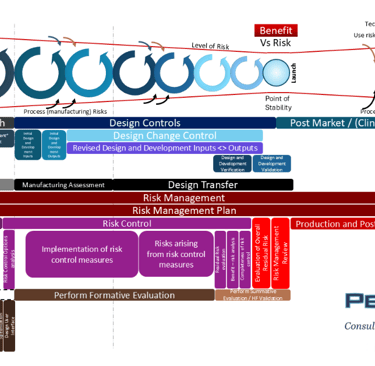
The final version of the new risk diagram - “big screen version” , which I will detail over the coming newsletters.
I’m hoping you’ll stick with me for this diagram-building and I encourage comment and discussion.
*note, nowadays I consider the diagram to be fully applicable to Combination Product development as well.